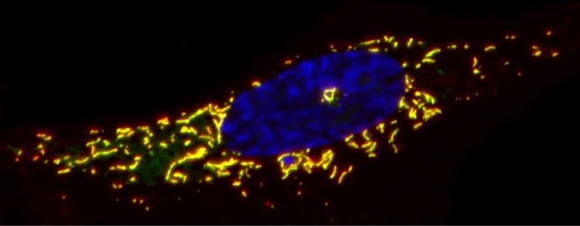
Research Summary:
Chromosomes are the fundamental structural units of our genome. Understanding their basic biology and how their mismanagement leads to disease is the main focus of the Sfeir lab. The scientific questions we address cover three major areas of research:
1 - Telomere maintenance and dysfunction:
A) Telomere elongation during embryonic development: A major goal of the Sfeir is to understand the mechanism(s) underlying telomere length regulation, which is critical for cellular survival. We are particularly interested in uncovering the pathway(s) that resets telomere length during embryonic development, a key stage that replenished telomere reserves for the adult. The activation of telomerase in the cells of inner cells mass is a crucial step, although its molecular underpinnings are largely unknown. We mapped hTERT enhancers and showed that transcriptional control does not fully account for the difference in hTERT mRNA levels between pluripotent and differentiated cells (Penev et al, biorxiv 2020). Instead, we made a striking observation that telomerase regulation is primarily governed by alternative splicing that involves hTERT exon-2. Furthermore, we implicate the splice co-factor SON in this hTERT splice switch during pluripotency and identify a patient carrying a SON mutation and suffering from dyskeratosis congenita as a result of short telomeres. Altogether, our study reveals that hTERT splicing is key developmental switch for telomerase that can lead to debilitating disease when defective. Our findings hold therapeutic implication both in the context of cancer treatment as well as boosting regenerative capacity of cells.
B) Telomere dysfunction and cancer: The G-richness and highly repetitive nature of telomeric DNA pose major challenges to the DNA replication machinery. In the lab, we recently highlighted a role for the telomere binding protein, POT1, in assisting the replisome when copying telomere repeats (Pinzaru et al., Cell Reports 2016). Notably, mutations in POT1 have been associated with several types of human cancers, including chronic lymphocytic leukemia (CLL), cutaneous T cell lymphomas (CTCL), and melanoma. We found that perturbations in POT1 (deletions and mutations) triggers telomere replication stress leading to profound telomere dysfunction. Together with the Lazzerini-Denchi lab (NIH), we showed that the loss of POT1 in the common lymphoid progenitor cells (CLPs) promotes the formation of thymic lymphomas in mice. Interestingly, we found that proliferation of cancer cells lacking POT1 is enabled by the attenuation of the ATR kinase pathway, highlighting a novel means by which cancer mutations bypass cell cycle checkpoint. In a subsequent study we interrogated the genetic susceptibilities of cells carrying POT1 mutations and identified a role for the nuclear pore complex in resolving telomere replication defects (Pinzaru et al., Genes & Dev 2020).
C) A surprising link between telomeres and metabolism: In addition to addressing the role of telomeres in cancer, my lab found that mice lacking the telomere binding protein Rap1 suffer from metabolic dysregulation (Yeung et al., 2013). Rap1 is a major telomere binding protein that also acts as a transcriptional regulator controlling adipogenesis. We highlighted a role for Rap1 in protecting from late-onset obesity. Obesity and its comorbidities are at the heart of public health concern in the 21st century and our work on Rap1 probes a unique link between aging and metabolic diseases.
2 - Investigating error-prone DNA repair
A major goal of the lab is to understand the mechanistic basis of Micro-homology mediated Eend-joining (MMEJ), a poorly understood yet highly error-prone repair pathway that drives chromosomal rearrangements in tumors. With this approach in hand we recently identified the promiscuous DNA polymerase theta (Polθ) as a key factor that introduces random nucleotides during MMEJ-mediated repair. Interestingly, we found that mutagenic repair by Polθ acts as a salvage pathway in the context of homology directed repair (HDR) defective tumor and allowing the survival of BRCA-mutated breast and ovarian tumors (Mateos-Gomez et al., Natture 2015). As a result, Polθ emerged as a compelling target for cancer treatment. Polθ is unique in that it is the only eukaryotic polymerase that also has a helicase domain. Work from our lab implicated Polθ –Helicase activity in counteracting RPA to decide on the fate of DSB in mammalian cells (Mateos-Gomez et al., NSMB 2017). In addition, we are currently investigating the function of Polθ in driving genetic diversity in normal cells as well as cancer cells and address the fundamental question related to the advantage of a highly error-prone pathway of repair and its role in genome plasticity.

3 - Investigating mitochondrial genome instability
In recent year, we decided to adopt our knowledge of studying nuclear DNA stability to the specifics of the mitochondria, and address outstanding questions related to mtDNA replication and repair.
To study mtDNA replication, we developed a novel and powerful single-molecule approach to examine mtDNA replication in vivo, and successfully applied this method to elucidate the mode of mtDNA replication, which has been a matter of controversy for two decades (Phillips et al., Mol Cell 2017). We also defined and for the first time the cause of the common deletion, an mtDNA abnormality that is central to numerous pathologies and aging. Our findings, together with the unique approaches we developed have been very well received by the mitochondrial community. More importantly, they provide a strong foundation for our future work, which aims to address questions related to the physiological impact of mtDNA lesions, including breaks and deletions.
Recently, we investigated mitochondrial DSBs (mtDSBs) using mitochondrial TALEN (mTLNs) that we engineered to induce breaks at specific loci within human mtDNA (Tigano et al., Nature 2021). Our work revealed that unlike the nucleus, mitochondria lack robust DSB repair capacity. Instead, broken mitochondrial genomes are predominantly degraded. Subsequent replication of intact mtDNA repopulates mitochondrial genome pool and reestablishes the appropriate copy number. Furthermore, we observed that shortly after mtDSB formation, mammalian cells trigger a type I interferon response as a result of mitochondrial herniation and escape of mitochondrial RNA to the cytoplasm.